The EU Pharmaceutical Package: Impact on Orphan Medicinal Products
In our article series, “A Deep Dive into the EU Pharmaceutical Package Changes,” we introduce the proposed changes in the new Pharmaceutical Package. This article reviews the proposed changes for data protection related to orphan medicinal products (OMPs) and is a continuation of the topic “Insight into Regulatory Protection Changes.”
What is an OMP according to the European Medicines Agency (EMA)?
An OMP is a medicine for the diagnosis, prevention, or treatment of a life-threatening or chronically debilitating condition that is rare (affecting not more than five in 10,000 people in the European Union) or where the medicine is unlikely to generate sufficient profit to justify research and development costs.
These drugs are eligible to receive the Orphan Disease Designation (ODD) status which provides incentives such as protection competition once on the market.
What’s driving the proposed changes to OMPs?
In August 2020, the European Commission published its evaluation of the medicines for rare diseases and children legislation. According to this evaluation, even though major changes have happened for patients in the therapeutic landscape, high unmet needs (HUMNs) still remain.
Rare diseases currently affect 30 million people in Europe and 300 million worldwide. However, 95% of rare diseases have no authenticated treatment, and only 28% of the OMPs target rare diseases without any treatment. 70% of rare diseases occur only in children, yet only 12% of treatments with an ODD target diseases affecting children. Between 2000 and 2019, 67% of ODD treatments targeted the same three disease areas: blood/blood forming organs, antineoplastic and immunomodulating agents, and dermatology.
The lack of effective treatments and HUMNs for rare diseases, particularly in pediatrics, is the major driver behind the European Commission’s proposed changes to OMP regulations.
What are the proposed changes?
The new EU Pharmaceutical Package contains a proposal for a new “master” Regulation, merging the regulation OMP No 141/2000 with the Commission Regulation No 726/2004 and the Pediatric Regulation No 1901/2006.
Today, the OMP market exclusivity is ten years, extending to 12 years if a pediatric investigation plan (PIP) is completed. A PIP ensures that the necessary data are generated through studies in children to support the authorization of a medicine for children . All applications for marketing authorization for new medicines must include the results of studies as described in an agreed PIP, unless the medicine is exempt because of a deferral or waiver.
With the current regulation, the Marketing Authorization Holder (MAH) can benefit from a six-month supplementary protection certificate (SPC) when they have fully implemented the measures of the PIP.
SPC is an intellectual property right that extends the patent right’s 20-year protection.
Patent and Extension of Protection

Under the new proposal, market exclusivity is shortened to nine years, with possible extension to 13 years if a panel of criteria is met. However, PIP completion will no longer be part of the market exclusivity extension criteria, but the six-month SPC extension for PIP completion remains in place.
Concerning well-established OMPs, the market exclusivity is reduced to five years with no possibility of extension. A well-established use is when an active ingredient of a medicine has been used for more than ten years and its efficacy and safety are established. In such cases, application for marketing authorization may be based on results from scientific literature.
The “significant benefit” definition of an OMP has been updated. The current definition is “a clinically relevant advantage or a major contribution to patient care” (Commission Regulation (EC) 847/2000 Article 3). The proposed definition is that orphan drugs benefit “a substantial part of the target population” (Commission Regulation (EC) 2023/0131 (COD) Article 2)
Other new OMP criteria include:
- The disease must maintain a prevalence of 5/10,000, with in-market products demonstrating the updated significant benefit.
- If the product does not meet the prevalence criteria, the European Commission can supplement with delegated acts for conditions that need orphan drugs.
- ODDs would be valid for a period of seven years. Extension might be possible if a sponsor provides evidence that a clinical trial is under way. However, the designation will expire when the product receives a marketing authorization.
Who benefits from the proposed changes?
Companies developing products in orphan disease and fulfilling the criteria below will be eligible for the maximum of 13 years market exclusivity:
- Addressing a HUMN (+one year)
- Launching the product in all 27 Member States within two or three years from marketing authorization, (+one year)
- Providing two new orphan indications (+two years in total)
The new regulation takes a modular approach that introduces a large range of protection periods varying between nine and 13 years depending on the above factors.
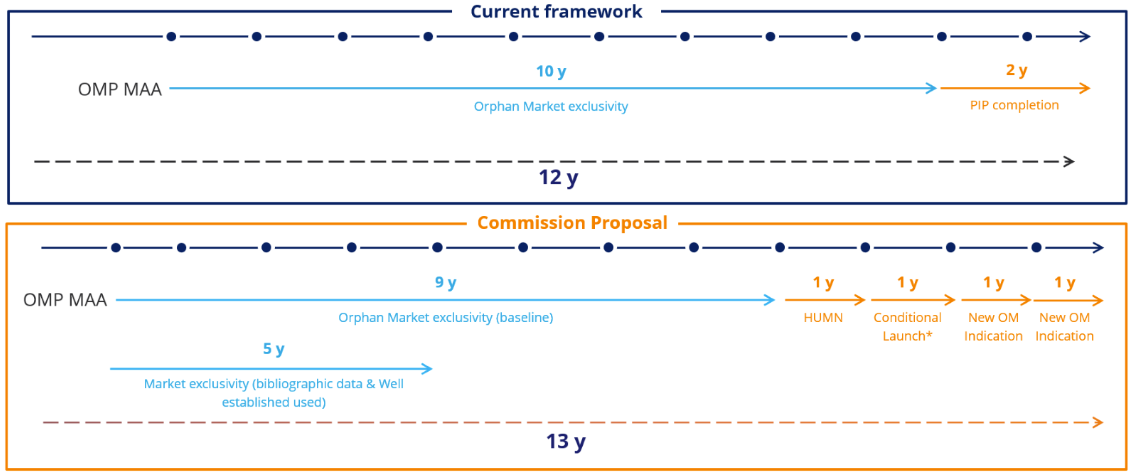
Orphan exclusivity: Current framework vs. new pharmaceutical package
Y: Year, HUMN: High Unmet Medical Need, OM: Orphan Medicine, PIP: Pediatric Investigation Plan
Case Study

How does the new EU Pharma Package compare to US regulations?
The orphan market exclusivity in the US regulation is seven years and six months from FDA approval with all the conditional extensions.
EU regulations will remain favorable in comparison, with from nine up to 13 years of market exclusivity depending on the conditions fulfilled.
What are the challenges with the new proposal?
While the general objectives of the proposed EU Pharmaceutical Package are necessary, several topics remain confusing, especially the interpretation of some definitions such as HUMN.
OMP framework
The additional “significant benefit” (Commission Regulation 2023/0131 (COD), Article 2) definition criterion is already part of the working guideline currently in use by the EMA, but the formal inclusion as part of the updated Regulation has the potential to exclude drugs that would have previously qualified. The notion of “substantial part” is difficult to define and makes the system even more unpredictable.
Furthermore, the proposal that the ODD expires at the point of marketing authorization or after a seven-year period is concerning. The potential benefit of this change is not highlighted, and it will have a significant negative impact on national legislation where incentives are tied to therapy designation. This change could have dire consequences at the national level for OMP developers.
HUMN
Some treatments may respond to the HUMN definition to obtain an additional year of exclusivity to the baseline. However, the definition is vague, notably in the case of concepts such as “exceptional therapeutic advancement” or “meaningful reduction in disease morbidity or mortality” (Commission Regulation (EC) 2023/0131 (COD) Article 2).
HUMN is not a fixed concept and implementing a statutory definition in the legislation will compromise the value of innovation. Also, the definition does nothing to solve the main problem, namely, the lack of knowledge on rare diseases.
Launch conditionality
The need to launch a product in all 27 Member States within two or three years from the marketing authorization to obtain an additional year of OMP marketing exclusivity seems complicated, if not unfeasible, for small and medium-sized entities (SMEs).
However, the Commission is enabling some leeway by allowing certain developers to have three years to launch. Those developers are the not-for-profit entities, the SMEs (as in Commission Recommendation 2003/361/EC), or developers that have received fewer than five centralized marketing authorizations.
For the benefit of these developers and for patients awaiting innovative medicines, the hope is that that the European Commission will consider extending this requirement to four years.
New indication
In the new EU Pharmaceutical Package regulation, “the first two new indications of an OMP will be rewarded with one year of exclusivity each” (Commission Regulation (EC) 2023/0131 (COD) Article 2).
Unfortunately, the regulation does not include a precise definition of a “new indication” (Commission Regulation (EC) 2023/0131 (COD) Article 2). Furthermore, the regulation states that an ODD ceases to be valid once the OMP has obtained a marketing authorization. It is unclear if the current regulation in which a sponsor cannot have orphan and non-orphan indications under the same license remains valid.
Conclusion
This new European Commission proposal may introduce uncertainty for SMEs developing innovative therapies for rare diseases. The window for industry stakeholders to submit feedback to the EC closed August 1, 2023, and the proposed EU Pharmaceutical Package remains under discussion until final adoption by the European Parliament and EU Council.
More EU Pharmaceutical Package Resources
Subscribe to our newsletter for the latest news, events, and thought leadership